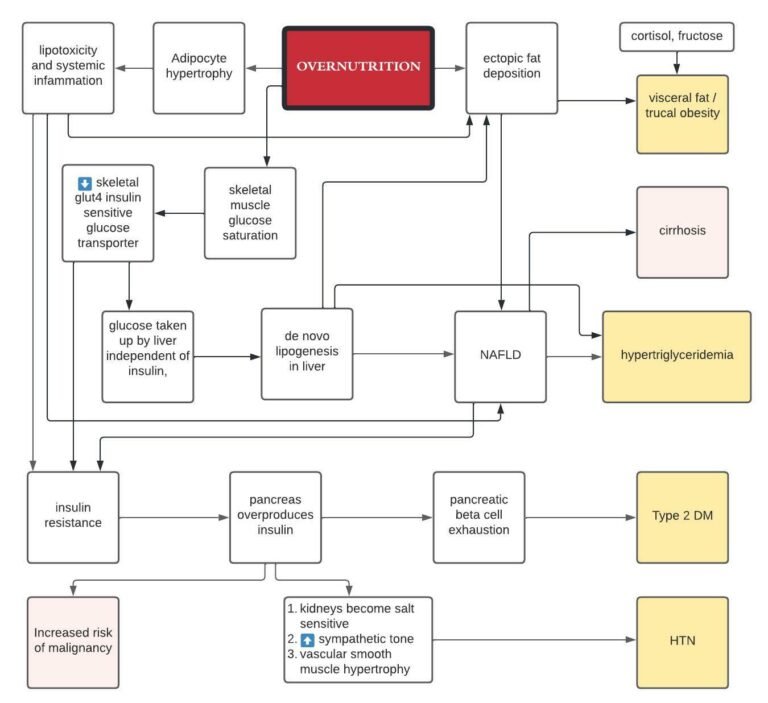
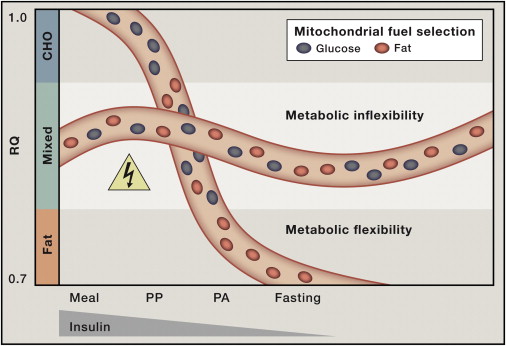
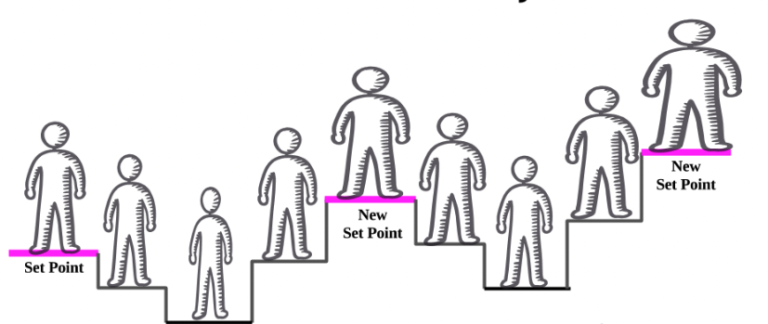
Circadian interventions
We have internal rhythms that align with the rotation of the planet. Most famously, there is the rhythmic secretion of melatonin at night and a spike of cortisol in the morning. These and myriad other oscillations are controlled by aptly named clock genes that are present in every cell of the body. Clock genes work…