Why do we have to die?
If we are to understand aging, the primary question is why should there be aging and death in the first place? Why is it almost universal in living organisms and what evolutionary purpose, if any, might aging and death serve? First let’s clarify that I don’t have any formal education in evolutionary biology, I think it’s a fascinating field and I’m frustrated by my status as a dilettante. Yet I proceed in the spirit of an amateur and offer my hypothesis.
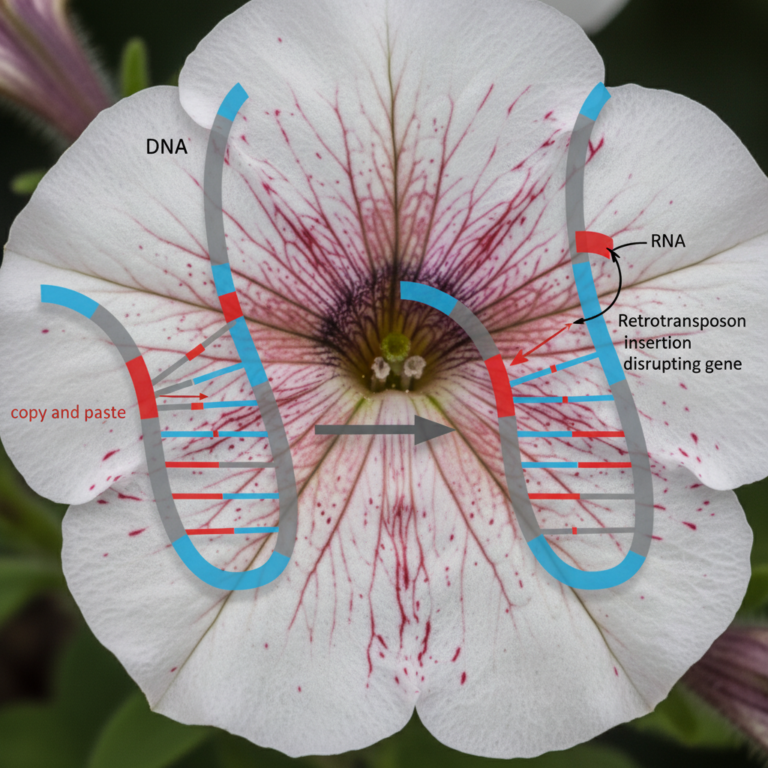
Aging really looks programmed. Take the Pacific salmon—he swims upstream, spawns, and dies on a reliable schedule. Humans lose muscle, go through menopause, develop male-pattern baldness, and accumulate frailty in stereotyped sequence. The epigenetic clocks developed by Horvath and others tick along a remarkably consistent trajectory across individuals—the methylation pattern of a sixty-year-old liver looks reliably different from that of a thirty-year-old liver, in ways that are not random.
If aging were simply the accumulation of damage, we would expect it to look noisy and idiosyncratic. Instead it looks more like development. It looks like puberty: a sequence of events unfolding on a clock. Are there bald adolescents? Not so much. The question is whether this appearance of programming reflects something real, or whether it is an illusion produced by deeper, non-programmatic forces.
Yet the mainstream view of aging is hostile to a program. Aging, in the conventional perspective, is a manifestation of neglect rather than intent.
Mutation accumulation (Medawar, 1952) holds that harmful mutations expressed late in life are weakly selected against, because most organisms in the wild are killed by predators, disease, or accidents long before those mutations matter. Over many generations, late-acting harmful mutations become part of the genome simply because they cannot get selected against prior to reproduction. This explains some features of aging—particularly the late-life rise in conditions caused by recessive variants—but it does not predict the choreographed quality of the aging process.
Antagonistic pleiotropy (Williams, 1957) goes further. It proposes that some genes are actively selected for despite causing aging, because the same genes confer reproductive advantages early in life. Hyperfunction that primes an organism for reproduction ultimately speeds aging, burning the organism out. Williams’s framework can explain a great deal of what looks programmed about aging, because under antagonistic pleiotropy, a great deal of aging really is the predictable downstream consequence of genes that have been actively selected for.
Disposable soma (Kirkwood, 1977) extends this argument: the cell must allocate finite resources between reproduction and somatic maintenance. An organism that spent everything on DNA repair would be outcompeted by one that spent more on offspring. Aging emerges as the consequence of an evolved allocation strategy.
These three theories are usually presented as a package, but they are distinct and partly opposed, and together they explain much of what we observe. I want to concede this clearly: most of what looks programmatic about aging is probably accounted for by these mechanisms, particularly antagonistic pleiotropy. Anyone proposing something stronger needs to explain what AP cannot.
Three features of aging give me pause about whether antagonistic pleiotropy is the whole story.
- The precision of the program. The epigenetic clocks tick at a consistent rate across individuals of the same species, and at very different rates across species. A mouse and a human have nearly identical genomes at the level of base pairs that matter for cellular machinery, yet the mouse ages thirty times faster. The difference is regulatory and developmental, and it looks tuned. Antagonistic pleiotropy can produce aging, but it is not obvious why it would produce aging on a clock.
- Continuity with development. The molecular machinery that drives development—Hox genes, hormonal cascades, the epigenetic regulatory apparatus—does not switch off at sexual maturity but rather it keeps running. The same systems that orchestrate puberty may also orchestrate menopause, sarcopenia, and immunosenescence. If aging were just the accumulated cost of pleiotropic genes optimized for early reproduction, we would not necessarily expect it to look like a continuation of the developmental program. Yet it does.
- Sequence and uniformity. Within a species, the order in which things fail is conserved. Hair grays before joints fail before cognition declines. This is not what stochastic damage produces. The sequential conserved nature of aging makes it feel like programatic development, and the mechanism for the program is likely epigenetic.
Now suppose that, on top of antagonistic pleiotropy, there is some additional selection pressure favoring finite lifespans because populations of finite-lived organisms adapt faster than populations of long-lived ones. More generational turnover means more iterations of natural selection per unit time, which means faster accumulation of beneficial variants, which means a better chance of tracking environmental change.
In environments where change is rapid, such as the Cambrian explosion and the post-permian recovery, the early Cenozoic period and most recently, the Pleistocene epoch, there is accelerated evolutionary change. This is shown in the fossil record. Rapid evolution is advantageous during these periods of instability because organisms with accelerated evolutionary change will be better able to specialize to new niches and outcompete and displace those who evolve slowly. A population that adapts twice as fast can outcompete and displace a population that adapts half as fast, even if the latter has more reproductive years per individual.
But then there is the problem of the cheater. Imagine a population of programmed-death organisms. A single mutant arises whose program fails and it becomes the immortal cheater. In the very next generation, the cheater reproduces while the rest of the population dies on schedule. The cheater’s offspring inherit the broken program. Within a few generations, the cheater lineage dominates. But what I’m saying is that this would need to be balanced against the evolutionary fitness accrued by shorter lifespans. In other words, whatever evolutionary benefit of more offspring accrued from a longer cheating life would be balanced by better evolutionary fitness from more cycles of natural selection due to shorter life. So the cheater genotype ultimately loses out to the short-lived genotype.
This is the end of my brief career as an evolutionary biologist.