Autophagy
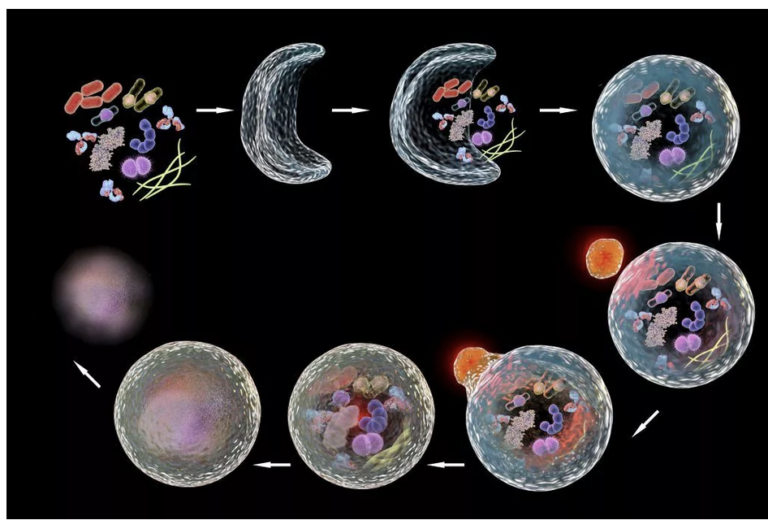
Autophagy (literally “self-eating”) is the body’s way of cleansing cells by recycling old or damaged components and is a process that appears to have a strong relationship to preventing disease and aging. More technically, it is a highly regulated lysosome-dependent catabolic program used by the body to clear dysfunctional proteins, organelles and other structures that…
